Abstract
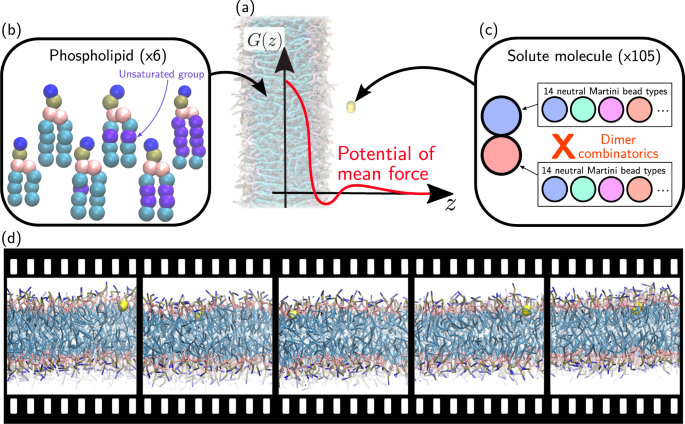
The permeation of small-molecule drugs across a phospholipid membrane bears much interest both in the pharmaceutical sciences and in physical chemistry. Connecting the chemistry of the drug and the lipids to the resulting thermodynamic properties remains of immediate importance. Here we report molecular dynamics (MD) simulation trajectories using the coarse-grained (CG) Martini force field. A wide, representative coverage of chemistry is provided: across solutes—exhaustively enumerating all 105 CG dimers—and across six phospholipids. For each combination, umbrella-sampling simulations provide detailed structural information of the solute at all depths from the bilayer midplane to bulk water, allowing a precise reconstruction of the potential of mean force. Overall, the present database contains trajectories from 15,120 MD simulations. This database may serve the further identification of structure-property relationships between compound chemistry and drug permeability.